


INTRODUCTION
Over the past two decades,the compounds of ar-senic have been studied experimentally and theo-retically because they have been used as pigmentssince ancient times and used as compo-nents tomodify the mechanical properties of lead andcopper alloys and to eliminate unwanted colorationof glasses in modern industries.1,2 The arsenicclusters have played prominent roles not only inthe early development of both forensics and chemo-therapy but also in many diferent fields of modernscience,such as solid-state physics,biochemistry,physical chemistry,surface phenomena,and catal-ysis.1-17 The knowledge of the fundamen-tal prop-erties,such as the ground and low-lying electronicstates of neutral and charged arsenic clusters,elec-tron a区nities (EAs),and ionization potentials (IPs),should lead to a more complete understanding oftheir role in many diferent fields.With this motiva-tion,we have carried out a detailed study of thestructures and properties of neutral and chargedarsenic clusters by means of the higher level of theGaussian-3(G3)theory.18,19
There have been previous experimental studieson arsenic clusters.For example,Bennett et al.12have measured the appea-rance potentials and iontranslational energies for the negative ions As-,
As2-,and As3-formed by dissociative resonancecapture of As4.Lippactal.1 have probed the EAs ofAsn-(n=1-5)using photoelectron spectra in 1998.In 2002,Zhai etal.4have studied the electronicstructures and EAs of Pn5-(Pn=P,As,Sb,and Bi)with photoelectron spectroscopy and ab initio cal-culations.Recently,Walter et al.5 have measuredthe binding energy and fine-structure splitting offthe As-using infrared photodetachment thresholdspectroscopy.For IPs,Bhatia and Jones20 havemeasured the IPs of As by optical spectroscopy.Using gas-phase charge-transfer reactions,Zim-merman et al.3 have determined the IPs of arsenicclusters(Asn,n=1-5).Using high-resolution photo-electron spectra and photoionization mass spec-trometry,the IPs of As2 and As4 have been mea-sured by Wang et al.21-23 and Yoo et al.,10 re-spectively.Brumbach and Rosenblatt24 have in-vestigated the vibrational modes of As4 with Ramanspectroscopy.
There are many diferent methods to study thestructures and properties of Asn clusters,especial-ly for small clusters withn≤6.For instance,Scu-seria25 studied the bond length,harmonic vi-brational frequency,and IP for As2 with the CCSDmethod.Balasubramanianet al.11 investigated theelectronic structure and potential energy surface ofAs3 and its positive ion using CAS-MCSCF,followedbyMRCI.Meieretal.26 exploredthestructures,
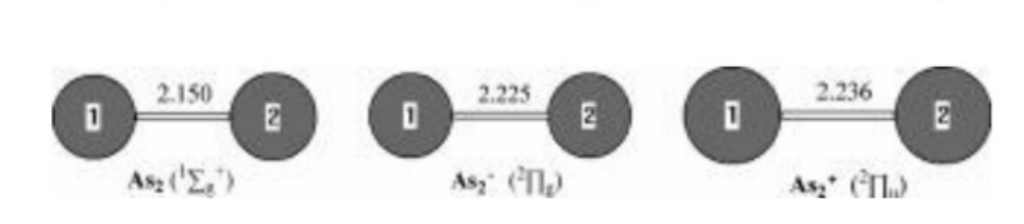
Figure 1.Neutral and charged As₂0-1-1)geometries optimized withthe MP2(full)scheme and 6-31G(d)bass set.The bond lengths are inangstroms.
stabilities,and ionization potentials of As2 and As4at the MRDCI level of theory.Warren et al.27 stud-ied the geometry of As6 with abinitioSCF-MO calcu-lations.BelBruno28 exploredthebonding propertiesof Asn(n=2-5)clusters using the B3PW91 scheme.Igel-Mannet al.29 probed the structures and verti-cal IPs of Asn(n=1-6)using SCF and CI methodswith energy-adjusted pseudopotentials.Zhao etal.8investigatedthestructuresand EAs of Asn (n =1-5)using various density functional theories(DFTs).For medium-sized Asn clusters,Ballone etal.30 explored the structures of Asn clusters up-ton=11usinglocal andnonlocal density functionals.Zhao et al.7 reported the study of structural andelectronic properties of Asn clusters in the sizerange of n≤28 using DFT with the PBE ex-change-correlation functional and all-electronbasis set of the double-numerical-plus-d-polar-iza-tion(DND)type.Guo6 investigated the geomet-rical and electronic properties of neutral andcharged Asn clusters with n=2-15 using theB3LYP/6-311+G(d)method.Besides these,Schae-fer and co-workers9 investigated the stability ofAsn clusters with n=2,4,12,and 20 using HF,MP2,CISD,and CCSD methods.Baruah etal.31explored the geometry,polarizabilities,and infraredand Raman spectra of fullerene-like arsenic cageswith n=4,8,20,28,32,36,and 60 by means ofGGA.
The objective of the present study is to apply ahigher level of G3 theory to the determination of theEAs and other properties of Asn (n=2-8)species.Of specific interest is (a)the com-parison of theelectron anities with the limited available ex-perimental results and (b)the predictions of otherproperties,including IPs,binding energies(BEs),and several dissociation energies(DEs).We wouldlike to establish reliable theoretical predictionsfor those arsenic clusters in the absence of experi-mental results and,in some cases,to challenge ex-isting experiments.
COMPUTATIONAL METHODS
All of the calculations at the extension of G3 theo-
ry18,19 have been performed using the Gaussian03 package.32 The combined G3 methods are abinitio calculations of molecular energies of com-pounds_containing first-,second-,and third-rowatoms.The average absolute deviation from experi-ment for the 423 reactions,including enthalpies offormation,ionization potential,electron a区nities,and proton a区nities,is 1.06 kcal/mol.19 Recently,we calculated the EAs of Sin clusters up to n=10using this G3 theory.The average absolute devia-tions from experiment are only 0.97 kcal/mol(0.042 eV).33
RESULTS AND DISCUSSION
As2 and Its Charged Molecules.The optimizedgeometries of the ground states of As2 and itscharged species are given in Figure 1.The As2 inits 12g+ground state has an experimental As-Asbond length of 2.103 A.34 The As-As bond length inAs2 is 2.150 A predicted by MP2(full)/6-31G(d),which overesti-mates the As-As bond distance by0.047 A.The best theoretical estimates for thisbond length are CCSD results of 2.107 A
Table 1.Zero-Point Vibrational Energy (ZPVE)Cor-rected Adiabatic Electron A区nities(EAs)and Adia-batic Ionization Potentials (IPs)for Asn(n=2-8)a
reported by Scuseria et al.25 and of 2.099 A re-ported by Shen et al.9 The CCSD deviations fromexperiment are only±0.004 A.
Anion As2-in its 2Ig ground state also has an ex-perimental As-As bond length of 2.239 A.1 TheMP2(full)/6-31G(d)results are 2.225 A,which arein fairly good agreement with the experimental values.The deviations from experiment are only0.014 A.We noted that the theoretical results ofBP86 and BLYP with the DZP++basis set are inaccord with the experi-mental values.8 The EAs ofAs2 are predicted to be 0.83 eV(the theoretical andexperimental EAs and IPs for Asn with n=2-8 areshown in Table 1),which agree with the experimen-tal values of 0.739 eV measured by photoelectronspectra.1
Cation As2+has a 2Iu ground state with alow-lying 22g excitedstate. Thislatter-stateis0.33eVhigher in energy atthe G3 level oftheory.The As-As bond length in the 2Iu state ispredicted to be 2.236A and in the 2∑g state is pre-dicted to be 2.143 A.The IPs with ZPVE correctioncalculated by G3 method are 9.87 eV for 12g+f21u and 10.18 eV for 12g+f2∑g+,which are in excel-lent agreement with experiment values of 9.893and 10.23 eV.21
As3 and Its Charged Molecules.Four minimastructures for neutral As3,two for anionic As3-,and one for cationic As3+are shown in Figure 2.Ascan be seen from Figure 2,the electronic states forthree isosceles triangle structures of neutral As3are,respectively,2B1,2A2,and 2A1 states.Ener-getically,the lowest-energy 2B1 structures aremore stable than that of 2A2 and 2A1 by 0.04 and1.77 eV,respectively.The fourth isomers,linearstruc-tures,have the 2Ig electronic state.Ener-getically,it is higher by 1.53 eV than that of the2B1 structure.Wenoted that the 2A2 state is morestable than the 2B1 by 0.02-0.05 eV in energy atthe various DFT and the CASSCF/MRSDCI levelsreported by Zhao et al.8 and Balasubramanianet al.,11 respectively.This means that both 2B1and 2A2 electronic states compete with each otherfor the ground state of As3.We also noted that theground state of As3 was a linear structure reportedby Guo.6
For the ground-state structure of anion As3-,Zhao et al.8 predicted it to bean isosceles trianglewith C2v symmetry and the 1A1 electronic state.Guo6 predictedittobeanequilateraltriangle
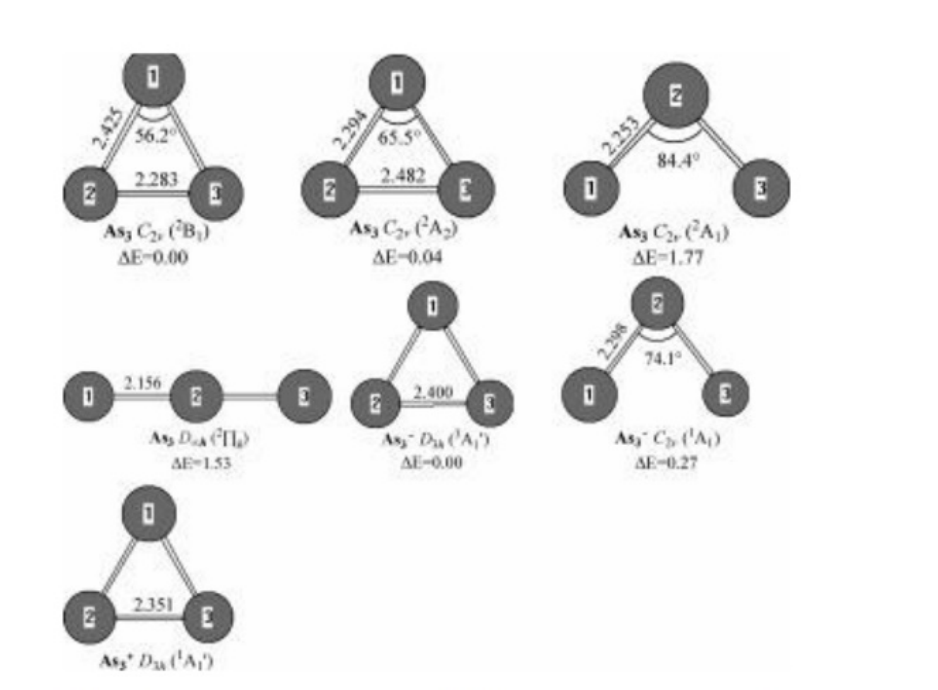
Figure 2.Neutral and charged As,0-1.+41)geometries optimized withthe MP2(full)scheme and 6-31G(d)basis set.The bond lengths andbond angles are in angstrons and degrees,respectively.The relativeenergies,△E,are obtained at the G3 level without ZPVE correctionand in eV.
with D3h symmetry and the 3A10 electronic state.At the G3 level,the 3A10 statestructureismor-establethanthe 1A1 stateby0.27eVin energy.TheEAs are predicted to be 1.80 eV for 2B1+3A10and 1.54 eV for 2B1+1A1 by means of the G3scheme.Lippaetal.1 have obtained the photoelec-tron spectrum of As3-with two major bands la-beled as A and B.Lippaetal.1 assigned band A asbeing due to the transition between the anion’sground state and the 2A2 ground state of As3 andband B as being due to the transition between theanion’s ground state and the 2B1 state of As3andconcludedthatthe EA ofAs3 was1.45±0.03eVandthat the energy gap between 2A2 and 2B1 iso-mers was 0.36 eV.In fact,the theoretical energygap between 2A2 and 2B1 isomers is only 0.05 eV,calculated with various theoretical methods,asmen-tioned above.Hence,the assignments forthe photoelectron spectrumof As3-are thought tobe unreasonable.Inthis case,we dare to reassignthe two major bands in the spectrum (see ref 1).Weassigned the band A as being because of thetransition between the 1A1 state of anion As3-andthe 2B1(or 2A2)ground state of neutral As3 andthe band B as being on account of the transitionbetween the 3A10 ground state of anion As3-andthe 2B1 (or 2A2)ground state of neutral As3.Thismeasured the experimental EA of As3 to be 1.45eVfor 2B1+1A1 and 1.81 eV for 2B1←3A10.Theenergy gap between 3A10 and 1A1 isomers forr As3-was measured to be 0.36 eV.All of these re-sults are close to the theoretical G3 values of 1.54,1.80,and 0.27 eV,respectively.This fact indicatesthat our assignment for the photoelectron spec-trum may be reasonable.
For the positively charged ionAs3+,theground-state structure is predicted to bean equilat-eral triangle with the 1A10 state.This result is thesame as the previous result reported by Balasubra-manianet al.11 and Guo.6 The G3 IP is predictedto be 7.33 eV,whichisinvery good agreement with-theoreticalvalueof7.1eV11 and experiment valuesof 7.49±0.13 and<7.32 eV.10
As4 and Its Charged Molecules.There have beena number of experimental and theoretical studieson As4.Among other reasons,this is because thetetrahedral As4 structure is the
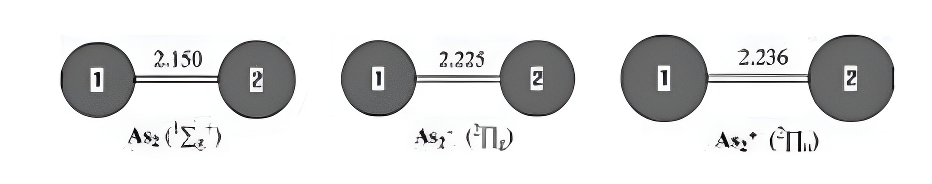
Figure 1.Neutral and charged As₂9-1,+1)geometries optimized withthe MP2(full)scheme and 6-31G(d)basis set.The bond lengths are inangstroms.
dominant form of elemental_arsenic under mostsublimation conditions. Previous investiga-tion2,6-9,29-31 revealed that As4 is a tetrahedralmolecule in its 1A1 ground state.The experimentalAs-As bond length was measured to be 2.435±0.04 A with a high-temperature gas-phase elec-tron diffraction technique.35 The G3 geometries ofthe ground states of neutral As4,anionic As4-,andcationic As4+are displayed in Figure 3.As can beseen from Figure 3,the As-As bondlength of 2.444Aevaluatedat the MP2(full)/6-31G(d)level is infairly good agreement with the experimental valueof 2.435 A taken from ref 35.
The ground-state structure of anion As4-pos-sesses_C2v sym-metry with the 2B2 electronicstate.The EAs are predicted to be 0.54 eV by meansof the G3 scheme.Zhao et al.8 calculated the EAstobe 0.49,0.50,0.62,and 0.65 eV using the BLYP,BHLYP,B3LYP,and BP86 method combined withthe DZP++basis set,respectively.Lippaetal.1 eval-uated an upper limit for the EAs of As4 to be <0.8eV using the photoelectron spectroscopy.The G3EAs are close to the DFT results8 and below theupper limit.1
For the positively charged ionAs4+,theground-state structure displays D2d symmetrywith the 2A1 electronic state.This result difersfrom the conclusion of the B3LYP/6-311+G(d).Atthe B3LYP/6-311+G(d)level,the ground-state
structure of As4+displays C2v symmetry with the2B2 state.6 The theoretical IPs are predicted to be8.65 eV,which are in very good agreement with ex-periment values of 8.63±0.1 eV.3 We noted thatWanget al.estimated the IPs for As4 to be 7.83 eVin the case of(1e)-1 by Jahn-Teller spectral simula-tion.22
The experimental EAs and IPs of As2-4 can be criti-cal bench-marks for testing the accuracy of the G3theoretical methods.For the six reactions,includ-ing two EAs(As2 and As3)and four IPs(two forAs2,one for As3,and one for As4),the average ab-solute deviation from experiment is only 0.05eV.As5 and Its Charged Molecules.There are a fewprevious theoretical studies on the structure of theground state of the As5 cluster with the DFTscheme.The DFT results showed that the low-est-energy isomes of As5 possess C2v symmetrywith the 2B1 state.6,8,30 The G3 result shown inFigure 4 is the same as the previous DFT conclu-sion.6,8,30
For the anionic As5-species,the lowest-energystructure shown in Figure 4 displays D5h symme-try with the 1A10 electronic state.These results arethe same as those of previous studies.4,6,8 Com-parison with the neutral As5 shows that there is asubstantial change in the geometry between theneutral and the anion.The G3 prediction of the EAsis 3.01 eV,which is the largest value among all ofthese reported in this paper.The reason for the un-usual electronic stability of anionic As5-is as-cribedto notonly the closed-shell nature of the As5-but also its aromatic charac-ter.4 The G3 predictedEAs of As5 are close to those estimated by theBHLYP,B3LYP,BP86,and BLYP schemes(the cal-culated values are 2.92,3.14,3.12,and 3.03 eV,respectively).8 It is a


larger diference between the experimental value of~1.71 and~3.51 eV.4 The EAs of~1.7 eV weremeasured by a photoelec-tron spectrum with2.497 eV photons.1 If the G3 value of 3.01 eV is re-liable,then the experimental value of~1.7 eV would be incorrect due to the deficiency of photon energy.Zhai et al.4 recorded two photoelectron spectrawith 4.661 and 6.424 eV photons.In light of thephotoelectron spectra of 4.661 eV photons,theEAs of 3.51 eV were determined.Although these ex-perimental values are in excellent accord withthose predicted by the B3P86 and LSDA(the B3P86and LSDAEAs are 3.55 and 3.54 eV,respective-ly),8 the B3P86 and LSDA results always over-estimate experimental values.36 In ref 36,Riens-tra-Kiracofe et al.reviewed the theoretical predic-tions of EAs with six DFT methods(B3LYP,B3P86,BHLYP,BLYP,BP86,and LSDA)and showed thatthe overestimated average errors of the B3P86 andthe LSDA forEAs of nearly 110 species are morethan 0.6 eV.All of these suggest that the G3 EAs of3.01 eV for As5 are reliable.
For the cationic As5+species,the lowest-en-ergy structure possesses C4v symmetry with the1A1 ground state.This result is the same as that ofprevious studies.6 Comparison with the neutralAs5 shows that there is a substantial changein the geometry between the neutral and thecation.The theoretical IP is predicted to be 6.68 eVat the G3 level.We noted that the deviation of thistheoretical IP from the experimental value of 7.95±0.1eV3 is1.27eV.No othertheoreticalorexperi-mentalIP is available for comparison.We expectfurther experimental studies and/or other higherlevel of theoretical calculations would be carriedout.
As6 and Its Charged Molecules.For neutral As6,previous studies7,27,30 showed that two low-lyingstructures are very close in energy.One is the benz-valene form with C2v symmetry and the 1A1 state,and another is a trigonal prism with D3hsymmetryand the 1A10 state,as shown in Figure 5a,b,respectively.Warren etal.27 reported thatthetrigo-nalprismislower in energythanthe benzvalene formby 0.24 eV at the HF/6-311G**level.Zhao et al.7also presented that the trigonal prism is morestable in energy than the benzvalene form by 0.13eV at the DFT-PBE/DND level.On the contrary,Ballone et al.30 reported that the benzvalene formis slightly more stable than the trigonal prism by0.06 eV at the LSD-DC level.Guo6 presented thatthe ground-state structure of As6 was a trigonalprism at the B3LYP/6-311 +G(d)level.We per-formed stil B3LYP/6-311+G(d)calcula-tions andfound that the trigonal prism is actually 0.08 eVhigher in energy than the benzvalene form.At theG3 level,energies of the benzvalene form and thetrigonal prism are nearly identical(benzvalene formis slightly less stable than the trigonal prism by
0.001 eV in energy).All of these indicate that thepotential surfaces of As6 are very flat,many iso-meric arrangements are possible,and accuratepredictions of equilibrium geometries require ad-vanced quantum mechanical investigations.

Figure 5.Neutral and charged As₆0-1-1)geometries optimized withthe MP2(full)scheme and 6-31G(d)basis set.The bond lengths are inangstroms.The relative energies,△E,are obtained at the G3 levelwithout ZPVE correction and in eV.
At the G3level of theory,the lowest-energy struc-ture of the negatively charged ion As6-is predictedto be the benzvalene form with C2v symmetry andthe 2A2 ground state (shown in Figure 5c).Thisresult is the same as the previous result reportedby Guo.6 The D3h trigonal prism of the 2A200state(shown in Figure 5d)is slightly higher thanthat of the ground state by 0.05eVin energy.Thetheoretical EAs are predicted tobe 2.08eV.No ex-perimental values are available for comparison.
For thepositivelycharged ionAs6+,threeisomersare-shown in Figure 5e-g.The structure(e),benzvaleneform,possesses C2v symmetry with the 2B1ground state.The isomer(f)displays Cs symmetrywith the 2A00 state.Energetically,it is higher thanthe ground structure(e)by 0.20 eV.The isomer(g)has C2h sym-metry with the 2Ag electronic state.
Guo6 reported that it was the ground state at theB3LYP/6-311+G(d)level of theory.At the G3 level,it is only the local minimum on the potential sur-face.Energetically,it is less stable thanthe groundstate(e)by0.79 eV.The theoretical IP is predicted tobe 7.97 eV.No experimental values are available forcomparison.
As7 and Its Charged Molecules.Two minimastructures for neutral As7,two for anionic As7-,and one for cationic As7+are exhibited in Figure 6.For neutral As7,one(Figure 6a)can be regarded ascapping the trigonal prism of As6 with an arsenicatom.Another(Figure 6b)can be regarded as cap-ping the benz-valene form of As6 with an arsenicatom.The structure of the capped trigonal prismdisplays C2v symmetry with the 2B1 state,and theisomer of the capped benzvalene form possessesCs symmetry with the 2A0 state.Energetically,theC2v structure is only more stable than the Cs by0.02 eV.It indicates that the
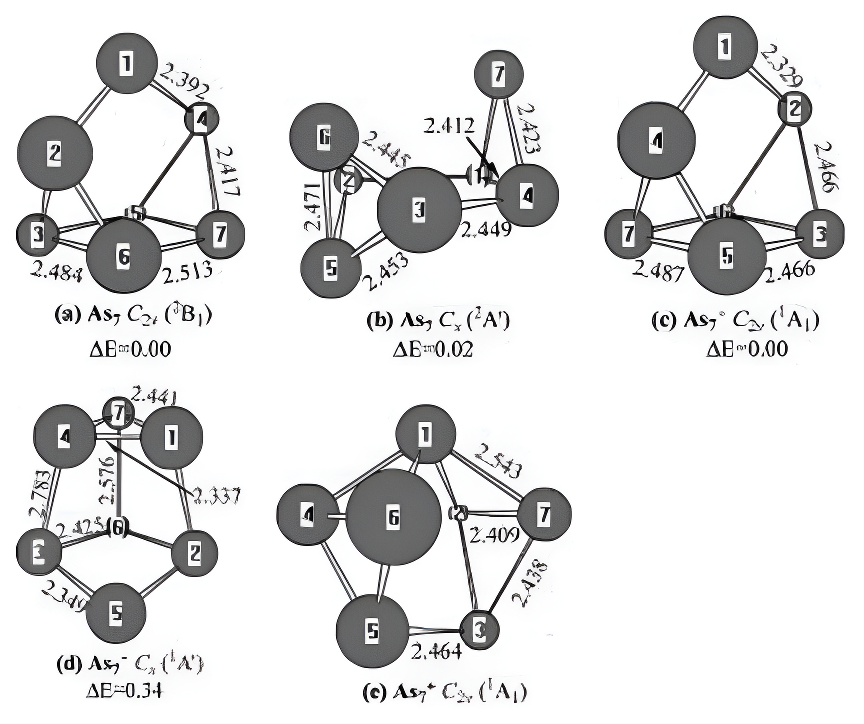
Figure 6.Neutral and charged AsO-1,+1)geometries optimized withthe MP2(full)scheme and 6-31G(d)basis set.The bond lengths are inangstroms.The relative energies,△E,are obtained at the G3 levelwithout ZPVE correction and in ev.
potential surfaces of As7 are very shallow.At theB3LYP/6-311+G(d)level of theory,the geometry ofthe capped trigonal prism is also the lowest-energystructure.6
Attaching an electron to the capped trigonalprism of neutral As7 results in an anionic As7-(Fig-ure 6c)with C2vsymmetry and the 1A1 electronicstate,which is the ground-state structure.Thisresult is the same as the conclusion ofB3LYP/6-311+G(d).6 Adding an electron to the
capped benzvalene form of As7 produces the anionAs7-(Figure 6d)with Cs symmetry and the 1A0electronic state.Energetically,the Cs structure isless stable than the C2v by 0.34 eV.The theo-retical EAs are predicted to be 2.93 eV.No experi-mental values are available for comparison.
For the positively charged ionAs7+,theground-state structure shown in Figure 6ehas C2vsymmetry with the 1A1 ground state.This result isthe same as the conclusion of B3LYP/6-311+G(d).6 The theoretical IP is predicted to be6.58 eV.No experimental values are available forcomparison.
As8 and Its Charged Molecules.For neutral As8,two minima structures are presented.The ditetra-hedron shown in Figure 7a can be regarded as at-taching two arsenic atoms to the benzvalene formof As6.The cagelike structure shown in Figure 7bcan be regardedas addingtwoarsenicatoms tothet-rigonalprismofAs6.The former has C2h symmetrywith the 1Ag ground state,and the latter displaysC2v symmetry with the 1A1 electronic state.Energetically,the C2h structure is more stablethan the C2v form by 0.09 eV.These results aredifferent from those of DFT outcomes.6,7,30 Atthe PBE and LSD levels of theory,the C2v cagelikeisomers are more stable than the ditetrahedron by0.32 and 0.57 eV in energy,respectively.7,30
For the negatively charged ion As8-,two so-mers are shown in Figure7c,d.Both structures(c)and(d)possess Cs symmetry with the 2A00 elec-tronic state.Energetically, the former,ground-state structure,is more stable than thelatter by 0.30 eV at the G3 level.This result difersfrom previous work by Guo.6 Guo6 reported thatstructure (d)in Figure 7 was the ground-statestructure for As8-.The theoretical EAs
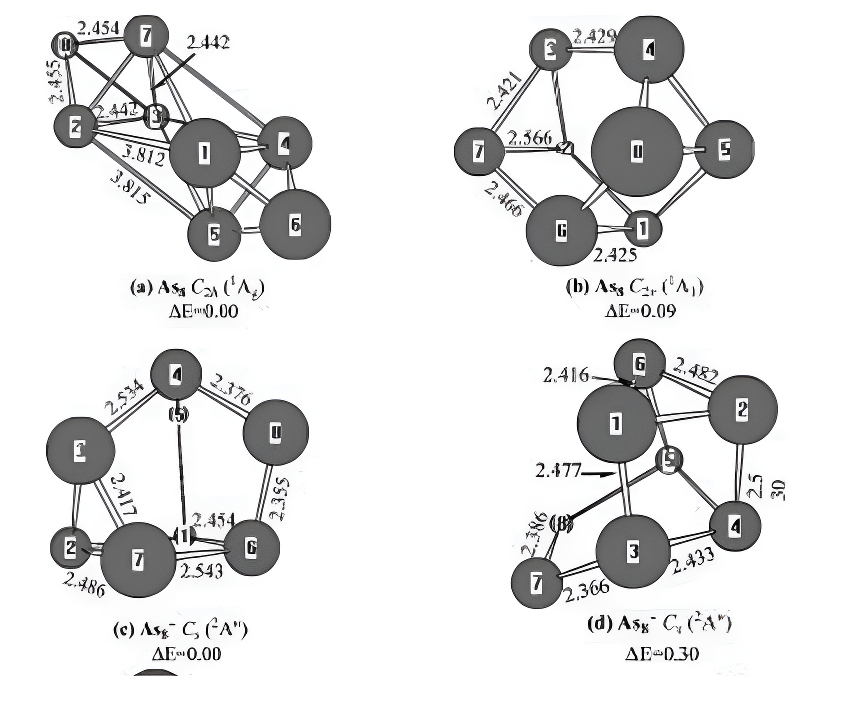
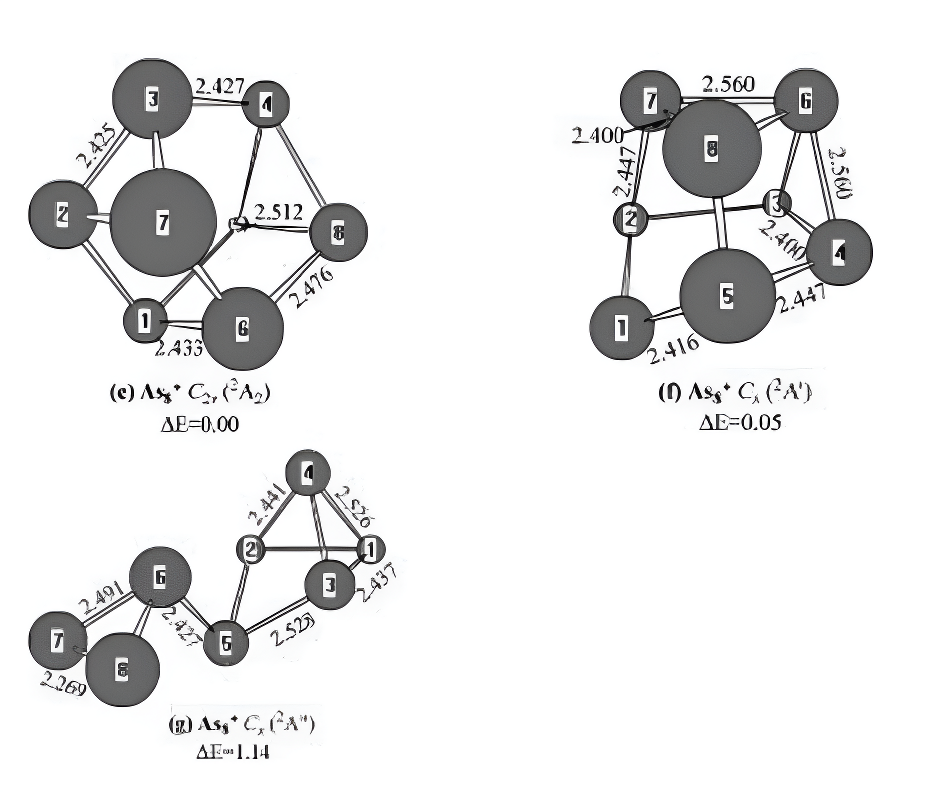
Figure 7.Neutral and charged As₂0-1,+1)geometries optimized withthe MP2(full)scheme and 6-31G(d)basis set.The bond lengths are inangstroms.The relative energies,△E,are obtained at the G3 levelwithout ZPVE correction and in eV.
are predicted to be 2.02 eV.No experimentalvalues are available for comparison.
For thepositivelycharged ionAs8+,threeisomersare-shown in Figure 7e-g.The cagelike structure(Fig-ure 7e)displays C2v symmetry with the 2A2ground state.The isomer of Figure 7f has Cs sym-metry with the 2A0 electronic state.Energetically,it is only less 0.05eV stable than the ground-statestructure.The isomer of Figure 7g possesses Cssymmetry with the 2A00 electronic state.Guo6 re-ported that structure(g)in Figure 7 was the groundstate of cationic As8+.However,it is less stablethan the 2A2 ground-state structure by 1.14 eVin energy.The theoretical IP is predicted to be7.65 eV.No experimental values are available forcomparison.
It is interesting to note that two types of structuralpatterns compete with each other for theground-state structure of neutral Asn starting fromn≥6.One type is derived from the benzvaleneform of As6,and another is derived from thetrigonal prism of As6.
Table2.Total Binding Energies(BEs)and Binding EnergiesPer Atom(BEPA)ofAsn(n=2-8)
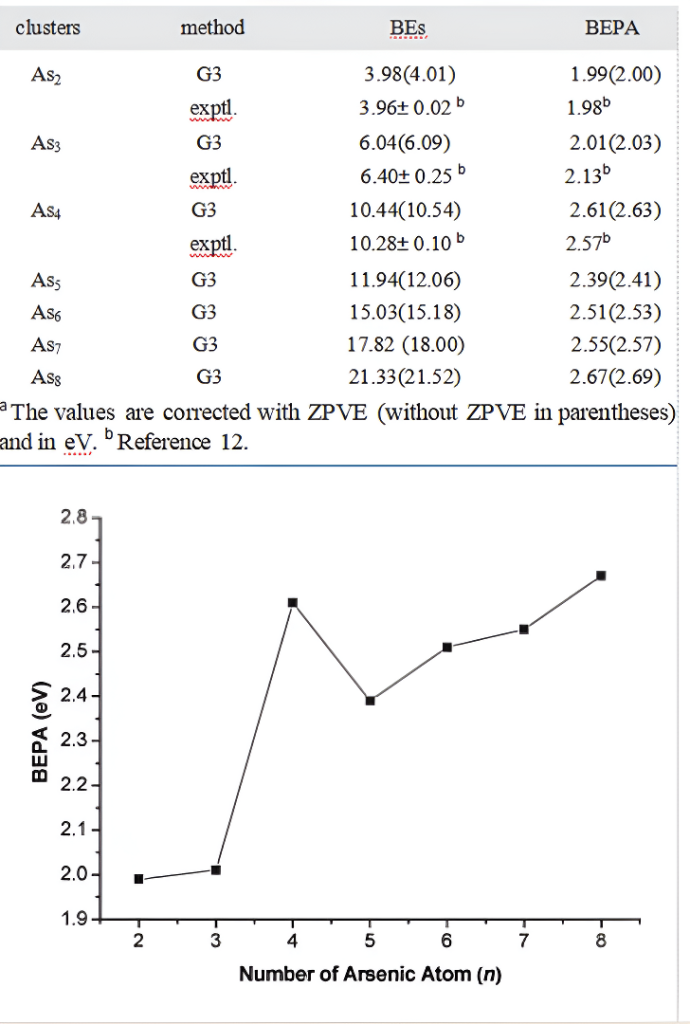
Figure 8.Binding energies per atom(BEPA)(eV)with ZPVE correc-tions versus the number of atoms n for Asn(n=2-8).
Binding Energies.TheBEs(defined as the energyrequired in the reaction Asn f nAs for Asn,that is,BE(Asn)=[nE(As)-E(Asn)])of Asn are evaluatedand listed in Table 2.The experimentally knownBEs are 3.96±0.02 eV for As2,6.40±0.25eV forAs3,and 10.28±0.10 eV for As4.12 The G3 valuesof 3.98 eV(As2),6.04 eV(As3),and 10.44 eV (As4)are in fairly good agreement with those of experi-ment taken from ref 12.The reliable BEs are pre-dicted with the G3 method to be 11.94 eV for As5,15.03 eV for As6,17.82 eV for As7,and 21.33 eVfor As8.
In addition to the total binding energies,it is alsoillustrative to consider the binding energies peratom(BEPA)in order to compare the stabilities ofdiferent clusters.The BEPA of Asn is also listed inTable 2.As can be seen from Table 2,the G3 BEPAvalues deviate -0.01 eV for As2,0.12 eV for As3
Table 3.Dissociation Energies(DEs)ofAsn(n=2-8)
dissociation pathways are listed in Table 3.As wecan seen from Table3,the smallest energy neces-sary to fragment the clusters into smaller pieces forAsn with n=5-8 corresponds to the process Asn fAs4+Asn-4.Such dissociation energies for As5-As8 at the G3 level are 1.49,0.61,1.34,and 0.44eV,respectively.
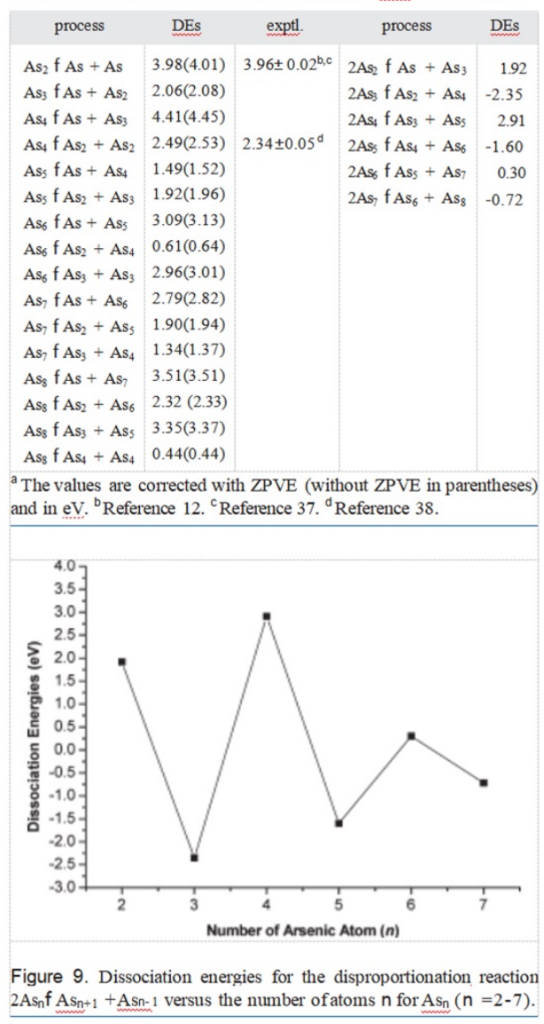
Other dissociation energies(defined as the energiesrequired forthedisproportionation reaction2Asn-fAsn+1 +Asn-1,thatis,DE(Asn)=[E(Asn+1)+E(Asn-1)-2E(Asn)])are also listed in Table 3 andshown in Figure 9.This gives a sensitive measure ofrelative stability.Figure 9 reproduced the conclu-
sion (the cluster size distributions exhibit strongeven-odd alternations)reported by Guo.6 Theeven-numbered clusters with n=2,4,and 6 aremore stable than the odd-numbered with n=3,5,and 7.The simple explanation is that theeven-numbered clusters are closed-shell,while theodd-numbered clusters are open-shellsystems.6Another measure of cluster stability is given by thedissociation energy,DE(Asn)=[E(Asn-1)+E(As)-E(Asn)]
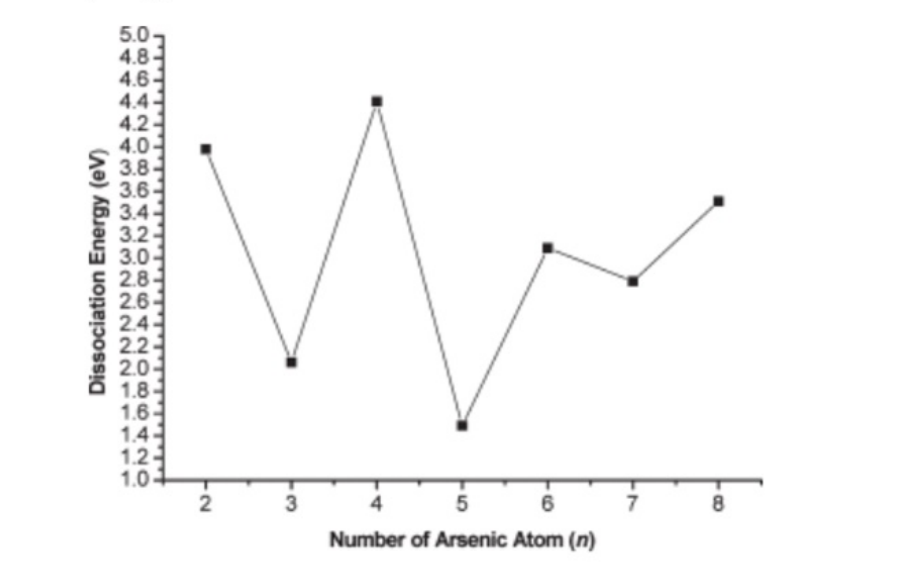
defined as the energies required for the reaction Asnf Asn-1+As.This is plotted in Figure 10.As can be seen from Figure 10,italso characterizes the even-numbered clusters as more stablethan the odd-numbered systems.
CONCLUSIONS
The structures and energies of neutral andcharged small Asn(-1,0,+1)(n=2-8)clusters havebeen systematically inves-tigated by means of theG3 schemes.The electron a区nities,ionization po-tentials,binding energies,and dissociation ener-gies have been calculated and compared with limit-ed experimental values.The results can be sum-marized as follows:(i)The potential surfaces ofneutral Asn clusters are very shallow,and twotypes of structural patterns compete with eachother for the ground-state structure of Asn with n≥6.One type is derived from the benzvalene formof As6,and another is derived from the trigonalprism of As6.(ii)In light of the G3 theoretical re-sults,we reassigned the photoelectron spectrum ofAs3-presented previously in ref 1.The experimen-tal electron a区nities of As3 were measured to be1.81 eV,not 1.45 eV.In terms of the G3 and the DFT results,we thought the experimental electrona☒nities of 3.51 eV for As5 taken from ref 4 to beunreliableThe reliable electron a区nities were pre-dicted to be 0.83 eV for As2,1.80 eV for As3,0.54eV for As4,3.01 eV for As5,2.08 eV for As6,2.93 eVfor As7,and 2.02 eV for As8.(ii)The G3 ionizationpotentials were calculated to be 9.87 eV for As2,7.33 eV for As3,8.65 eV for As4,6.68 eV for As5,7.97 eV for As6,6.58 eV for As7,and 7.65eV forAs8.These theoretical values of As2,As3,and As4are in excellent agreement with those of experi-mental results.(iv)Calculations of total bindingenergies and binding energies per atom were per-formed.The bind energies per atom were evaluatedto be 1.99 eV for As2,2.01 eV for As3,2.61 eV forAs4,2.39 eV for As5,2.51 eV for As6,2.55 eV forAs7,and 2.67 eV for As8.The As2,As3,and As4values,again,are in excellent accord with those ofexperimental results.(v)Several dissociation en-ergies were carried out to examine relative sta-bilities.The results characterized the even-num-bered clusters as more stable than the odd-num-bered systems.
We hope that our theoretical predictions will pro-vide strong motivation for further experimental in-vestigation of these im-portant arsenic clusters.
AUTHOR INFORMATIONCorresponding Author
*Fax: 86-471-6576145.E-mail:yangic@i-mut.edu.cn.
ACKNOWLEDGMENT
This work has been financially supported byGrants(Nos.40572149 and 40772162)from theNational Natural Science Foundation of China,bythe Foundation(No.2006CB202205)from the Na-tional Key Basic Research(973)Program,and byGrants(Nos.2007BAK24B01 and 2006BAB16B04)from the National Key Technology R&₆D Program forthe 11th Five-Year Plan of China.
REFERENCES
(1)Lippa,T.P.;Xu,S.-J.;Lyapustina,A.;Nilles,J.
M.;Bowen,K.H.J.Chem.Phys.1998,109,10727.
(2)Alcami,M.;M,O.;Y Chem.Phys.1998,108,8957.(3)Zimmerman,J.A.;Bach,S.B.H.;Watson,C.
H.;Eyler,J.R.
J.Phys.Chem.1991,95,98.
(4)Zhai,H.-J.;Wang,L.-S.;Kuznetsov,A.E.;Boldyrev,A.I.J.Phys.Chem.A 2002,106,5600.(5)Walter,C.W.;Gibson,N.D.;Field,R.L.;
Snedden,A.P.;Shapiro,J.Z.;Janczak,C.M.;Hanstorp,D.Phys.Rev.A2009,80,014501.(6)Guo,L.J.Mater.Sci.2007,42,9154.(7)Zhao,J.;Zhou,X.;Chen,X.;Wang,J.;Jell-inek,J.Phys.Rev.B 2006,73,115418.(8)Zhao,Y.;Xu,W.;Li,Q.;Xie,Y.;Schaefer,H.F.J.Comput.Chem.2004,25,907.(9)Shen,M.;Schaefer,H.F.J.Chem.Phys.1994,101,2261.(10)Yoo,R.K.;Ruscic,B.;Berkowitz,J.J.Chem.Phys.1992,96,6696.(11)Balasubramanian,K.;Sumathi,K.;Dai,D.J.Chem.Phys.1991,95,3494.(12)Bennett,S.L.;Margrave,J.L.;Franklin,J.L.;Hudson,J.E.J.Chem.Phys.1973,59,5814.(13)Xu,W.;Li,G.;Yu,G.;Zhao,Y.;Li,Q.;Xie,Y.;Schaefer,H.F.J.Phys.Chem.A 2003,107,258.(14)Kasalov,V.;Schaefer,H.F.J.Comput.Chem.2005,26,411.(15)Dong,C.;Yang,J.;Ning,H.;Li,C.J.Mol.Struct.:THEOCHEM 2010,958,26.(16)Gao,A.;Li,G.;Chang,Y.;Chen,H.;Li,Q.
J.Mol.Struct.:THEOCHEM 2010,961,88.
(17)Gao,A.;Li,G.;Chang,Y.;Chen,H.;Finlow,
D.;Li,Q.Inorg.Chim.Acta 2011,367,51.
(18)_Curtiss,L.A.;Raghavachari,K.;Redfern,P.
C.;Rassolov,V.;Pople,J.A.J.Chem.Phys.1998,
109,7764.(19)Curtiss,L.A.;Redfern,P.C.;Rassolov,V.;Kedziora,G.;Pople,J.A.J.Chem.Phys.2001,114,9287.(20)Bhatia,K.S.;Jones,W.E.Can.J.Phys.1971,49,1773.(21)Wang,L.S.;Lee,Y.T.;Shirley,D.A.;Balasu-bramanian,K.;Feng,P.J.Chem.Phys.1990,93,6310.(22)Wang,L.S.;Niu,B.;Lee,Y.T.;Shirley,D.A.;Ghelichkhani,E.;Grant,E.R.J.Chem.Phys.1990,93,6318.(23)Wang,L.S.;Niu,B.;Lee,Y.T.;Shirley,D.A.;Ghelichkhani,E.;Grant,E.R.J.Chem.Phys.1990,93,6327.(24)Brumbach,S.B.;Rosenblatt,G.M.J.Chem.Phys.1972,56,3110.(25)Scuseria,G.E.J.Chem.Phys.1990,92,6722.(26)Meier,U.;Peyerimhof,S.D.;Grein,F.Chem.Phys.1991,150,331.(27)Warren,D.S.;Gimarc,B.M.;Zhao,M.Inorg.Chem.1994,33,710.(28)BelBruno,J.J.Heteroat.Chem.2003,14,189.
(29)Igel-Mann,G.;Stoll,H.;Preuss,H.Mol.Phys.1993,80,325.(30)Ballone,P.;Jones,R.O.J.Chem.Phys.1994,100,4941.(31)Baruah,T.;Pederson,M.R.;Zope,R.R.;Beltrn,M.R.Chem.Phys.Lett.2004,387,476.(32)Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;Scuseria,G.E.;Robb,M.A.;Cheeseman,J.R.;Montgomery,J.A.,Jr.;Vreven,T.;Kudin,K.N.;Burant,J.C.;Millam,J.M.;Iyengar,S.S.;Toma-si,J.;Barone,V.;Mennucci,B.;Cossi,M.;Scalmani,
G.;Rega,N.;Petersson,G.A.;Nakatsuji,H.;Hada,
M.;Ehara,M.;Toyota,K.;Fukuda,R.;Hasega-
wa,J.;Ishida,M.;Nakajima,T.;Honda,Y.;Kitao,O.;Nakai,H.;Klene,M.;Li,X.;Knox,J.E.;Hrat-chian,H.P.;Cross,J.B.;Bakken,V.;Adamo,C.;Jaramillo,J.;Gomperts,R.;Stratmann,R.E.;Yazyev,O.;Austin,A.J.;Cammi,R.;Pomelli,C.;Ochterski,J.W.;Ayala,P.Y.;Morokuma,K.;Voth,
G.A.;Salvador,P.;Dannenberg,J.J.;Zakrzewski,
V.G.;Dapprich,S.;Daniels,A.D.;Strain,M.C.;Farkas,O.;Malick,
D.K.;Rabuck,A.D.;Raghavachari,K.;Fores-
man,J.B.;Ortiz,J.V.;Cui,Q.;Baboul,A.G.;Cli-ford,S.;Cioslowski,J.;Stefanov,B.B.;Liu,G.;Li-ashenko,A.;Piskorz,P.;Komaromi,I.;Martin,R.L.;Fox,D.J.;Keith,T.;Al-Laham,M.A.;Peng,C.Y.;Nanayakkara,A.;Challacombe,M.;Gill,P.M.W.;Johnson,B.;Chen,W.;Wong,M.W.;Gonzalez,C.;Pople,J.A.Gaussian 03,revision C.02;Gaussian,Inc.:Wallingford,CT,2004.(33)Fan,H.;Yang,J.;Lu,W.;Ning,H.;Zhang,Q.J.Phys.Chem.A2010,114,1218.(34)Huber,K.P.;Herzberg,G.Molecular Spectraand Molecular Structure.IV.Constants of DiatomicMolecules;Van Nostrand Reinhold:New York,1979.(35)Morino,Y.;Ukaji,T.;Ito,T.Bull.Chem.Soc.Jpn.1966,39,64.(36)Rienstra-Kiracofe,J.C.;Tschumper,G.S.;Schaefer,H.F.;Nandi,S.;Ellison,G.B.Chem.Rev.2002,102,231.(37)Hultgren,R.;Orr,R.L.;Kelley,K.K.Supple-ments to Selected Values of ThermodynamicProperties of Metals and Alloys (Arsenic);University of California:Berkeley,CA,August1970.(38)Murray,J.J.;Pupp,C.;Pottie,R.F.J.Chem.Phys.1973,58,2569.